GSE52917
|
| What
We Learned |
- This study demonstrated a surprisingly early occurrence of miRNA
fingerprints associated with highly penetrant ALS mutations, even decades before the likely onset of disease.
- Considering that earlier commencement of neuroprotective treatment
is plausibly more effective and that, according to the results, molecular pathophysiology
precedes the onset of disease for many years.
- The early, subclinical onset of molecular pathology
provides also a possible explanation for the failure to substantially
attenuate disease progression by disease-modifying treatment attempts
in ALS to date.
|
- Another prominent finding of this study are the
highly homogenous miRNA profiles in genetically-defined familial ALS patients
and pre-manifest mutation carriers.
- Moreover, 91.7% of the miRNAs downregulated in the mutation carriers overlapped with
altered miRNAs in familial ALS.
|
- Riluzole (the only approved disease modifying ALS
drug) is thought to delay degeneration of motor neurons primarily by a reduction of glutamate release and excitotoxicity
but has only a modest effect on survival when treatment is started after onset of disease.
|
- To date, mutations in over 20 different genes have been implicated
in ALS pathogenesis, with a hexanucleotide expansion in C9orf72 and mutations in the genes coding for
SOD1, TARDBP and FUS being the most frequent.
|
- Although familial ALS comprises about
10–15% of all cases, the majority of ALS is considered sporadic
without a known family history of ALS.
|
- In the ALS SOD1-G93A mouse model, for example, upregulation of
muscle-specific miR-206 was evident in plasma even before the onset of symptoms (Toivonen et al., 2014), and miR-206 was
shown to be involved in regeneration of neuromuscular synapses that slows down disease progression.
- Intraventricular delivery of an inhibitor of miR-155
(anti-miR-155), a miRNA that was upregulated in spinal cords of
SOD1-G93A mice and patients with sporadic ALS, could significantly extend survival and disease duration of ALS model mice.
|
- Patients were considered sporadic cases based on a negative family
history.
- Additionally, the two most frequent known causes of
familial ALS, namely mutations in the SOD1 gene and a hexanucleotide repeat
expansion in C9orf72, were excluded by Sanger sequencing
|
|
| What
We Did |
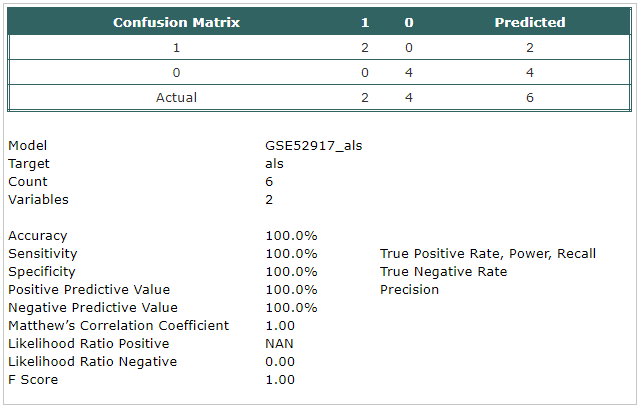
- A classification model has been built using Trainset.
The selected probes were:
- hsa-mir-1202_st
- hsa-mir-320a_st
|
- The model has been tested using Testset.
|
 |
| |
|
|